 Milano 18 Novembre 2022. Approvato da FDA il primo farmaco in grado di ritardare l’esordio del diabete di tipo 1, il suo nome è teplizumab.
Milano 18 Novembre 2022. Approvato da FDA il primo farmaco in grado di ritardare l’esordio del diabete di tipo 1, il suo nome è teplizumab.
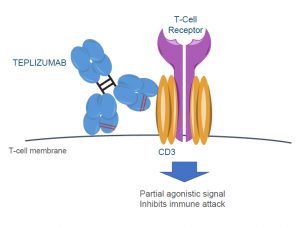
Cerchiamo di andare più in profondità per conoscere meglio questo nuovo farmaco. Il principio attivo denominato Teplizumab è un anticorpo monoclonale umanizzato della classe IgG1 che riconosce una parte di un complesso molecolare espresso sui linfociti T (CD3ε chain of the T-cell receptor complex), una classe di globuli bianchi coinvolta nella risposta immunitaria. Il farmaco non previene lo sviluppo del diabete di tipo 1 ma lo ritarda. Il farmaco è stato approvato da FDA il 17 novembre 2022 dopo una prima valutazione non positiva avvenuta nel Maggio 2021 per la necessità di completare alcuni dati di farmacocinetica e riassestare alcuni elementi della produzione.
Breve storia. La storia di questo farmaco è interessante. Discende da un precedente anticorpo monoclonale denominato OKT3 (muromonab) che è stato il primo anticorpo monoclonale approvato, nel 1986, dalle autorità regolatorie internazionali (FDA e EMEA) per l’uso clinico sull’uomo per ridurre la risposta immunitaria acuta nei soggetti sottoposti a trapianto d’organo. Il problema di OKT3 era che essendo di origine animale, determinava problemi importanti di tossicità, in particolare il rilascio massiccio di fattori infiammatori al momento della somministrazione (“cytokine release syndrome”) e la stimolazione del sistema immunitario contro il farmaco stesso. Per questo motivo l’anticorpo è stato modificato, tecnicamente “umanizzato”, sostituendo alcune parti proteiche originali con quelle degli anticorpi di origine umana, ottenendo teplizumab una versione meno immunogenica e all’inizio pensata per il trattamento del rigetto nei pazienti con trapianto di rene. Nel campo del diabete di tipo 1 è stato studiato in studi di fase 1-2 tra il 1990 e il 2005 e in 4 studi di fase 2-3 (AbATE, Delay, Protègè, Encore) tra il 2005 e il 2011 con lo scopo di proteggere al momento dell’esordio clinico della malattia le isole pancreatiche e la secrezione di insulina. Purtroppo gli studi di fase 3 fallirono in quanto l’efficacia ad un anno dall’utilizzo, valutata come fabbisogno insulinico e valori di emoglobina glicata, non fu diversa tra il gruppo trattato con il farmaco e il gruppo placebo. Per questo motivo teplizumab fu accantonato, anche se una parte del mondo scientifico riteneva che il fallimento degli studi di fase 3 fosse legato a problemi di disegno dello studio e ad un non corretto utilizzo. Il farmaco è stato poi “riesumato” nel 2011 all’interno di uno studio denominato TN-10 condotto da TrialNet, una rete dei principali ricercatori mondiali sul diabete di tipo 1, e finanziato dal National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) e JDRF. I risultati sono stati pubblicati sul New England Journal of Medicine e contemporaneamente presentati alla 79a sessione scientifica dell’American Diabetes Association del 2019. Lo studio TN-10 ha valutato teplizumab per la sua capacità di ritardare l’esordio clinico del diabete di tipo 1 nei pazienti in stadio 2 della malattia, definito dalla presenza di due o più autoanticorpi e disglicemia. Settantasei pazienti (teplizumab N=44, placebo N=32) sono stati arruolati con età compresa tra 8 e 49 anni, con il 72% di età inferiore ai 18 anni, e randomizzati a ricevere un singolo ciclo di 14 giorni di teplizumab o placebo mediante infusione endovenosa. Con un tempo di osservazione mediano di 51 mesi, la terapia con teplizumab ha comportato un ritardo di 25 mesi statisticamente significativo nello sviluppo della malattia. Nel marzo del 2021 è stato poi pubblicato un aggiornamento dell’andamento dello studio che mostrava un ulteriore ritardo all’ultima valutazione di 32.5 mesi nei soggetti trattati con teplizumab. Sulla base di questi dati è stata richiesta l’approvazione a FDA. Nell’aprile del 2019 è stato iniziato anche lo studio PROTECT per valutare l’efficacia del farmaco nel momento dell’esordio per prevenire la perdita di funzione delle isole pancreatiche il cui risultati sono attesi nei prossimi anni.
Chi può potenzialmente beneficiare del farmaco. I soggetti con età ≥8 anni, con almeno due auto anticorpi e che abbiano una condizione di “disglicemia” definita con uno dei seguenti parametri: una glicemia a digiuno ≥110 mg/dL e <125 mg/dL, una glicemia ≥140 mg/dL e <200 mg/dL a due ore dopo un test da carico orale di zucchero (OGTT) o un valore di glicemia ≥200 mg/dL al tempo 30, 60 o 90 minuti durante OGTT)
Come si utilizza il farmaco e quali sono i potenziali effetti collaterali. Il farmaco prevede la somministrazione intravenosa ogni giorno (tempo minimo 30 minuti) per 14 giorni consecutivi con una dose proporzionale alla superficie corporea. Gli effetti avversi associati al farmaco sono legati al suo meccanismo di azione e come per tutti i farmaci non sono irrilevanti. Negli studi il 14% dei trattati con teplizumab ha interrotto la terapia a causa degli effetti collaterali contro il 4% del gruppo controllo. Le reazioni avverse più comuni (>10%) sono state linfopenia, rash, leucopenia e cefalea. Le attenzioni principali nell’utilizzo del farmaco riguardano questi potenziali problemi:
- Sindrome da rilascio di citochine (CRS): la CRS si è verificata in pazienti trattati con teplizumab durante il periodo di trattamento e fino a 28 giorni dopo l’ultima somministrazione del farmaco. Per questo motivo si deve premedicare con antipiretici, antistaminici e/o antiemetici
- Diminuzione dei linfociti: negli studi clinici, la linfopenia si è verificata nel 78% dei pazienti trattati. Per la maggior parte dei pazienti, i livelli dei linfociti hanno iniziato a riprendersi dopo il quinto giorno di trattamento e sono tornati ai valori precedenti al trattamento entro due-quattro settimane dal completamento del trattamento e senza interruzione della dose.
- Reazioni di ipersensibilità: Nei pazienti trattati si sono verificate reazioni di ipersensibilità acuta comprendenti malattia da siero, angioedema, orticaria, rash, vomito e broncospasmo.
- Potenziali nuove infezione o riattivazione di infezioni come quella da Epstein Barr Virsu (EBV)
- Vaccinazioni: teplizumab può interferire con la risposta immunitaria alla vaccinazione e diminuire l’efficacia del vaccino per cui si devono somministrare tutte le vaccinazioni adeguate all’età prima di iniziare il trattamento
Disponibilità del farmaco in Europa. Il farmaco non è ancora disponibile in Europa e al momento quindi non è possibile accedere a questo trattamento. Sarà nostra premura seguire l’evoluzione delle approvazioni e la disponibilità reale del farmaco al momento opportuno.
“E’ un importante passo nel campo del diabete di tipo1 – spiega Emanuele Bosi, primario dell’Unità di Medicina Generale indirizzo diabetologico ed endocrino-metabolico all’IRCCS Ospedale San Raffaele da sempre in prima linea negli studi di prevenzione, nonché uno dei principali ricercatori della rete TrialNet – che segna una svolta scientifica e culturale importante in questa malattia. Da anni siamo in grado di predire la malattia in modo adeguato, ma non potevamo offrire un trattamento con lo scopo di ritardare lo sviluppo della malattia. Ora questa prospettiva si sta concretizzando sperando che presto si potrà anche prevenire e non solo ritardare. Nel frattempo dobbiamo sottolineare l’importanza di iniziare ad immaginare come individuare i soggetti che potrebbero beneficiare di questo farmaco, per esempio con campagne di screening basate sul dosaggio degli auto anticorpi”
“Le conseguenze di questa approvazione sono molto più ampie di quanto non si possa immaginare e non riguardano solo gli aspetti strettamente clinici – sottolinea Lorenzo Piemonti direttore del DRI di Milano e primario dell’unità di medicina rigenerativa e dei trapianti all’IRCCS Ospedale San Raffaele – si discuterà molto sul rapporto rischio beneficio nella pratica clinica, si dovrà cambiare l’attuale disegno standard degli studi di prevenzione con qualche difficoltà per gli approcci che prevedono strategie in cui l’azione del farmaco potrebbe risultare antagonista ma soprattutto spingerà ad identificare precocemente la malattia, prima dell’esordio clinico.”
Rimane anche da comprendere il problema sul costo e l’accessibilità al farmaco. “Non abbiamo ancora idea in Europa, non avendo ancora l’approvazione da parte di Ema (Agenzia Europea per i medicinali), ma il prezzo annunciato solo per il farmaco dal produttore in Usa prevede un costo per paziente di 193.900 dollari – prosegue Piemonti -. Questo costo è più del doppio di quanto le assicurazioni in Usa si aspettavano di pagare e molto più alto della soglia di 48.900 dollari stimata nel 2020, sotto la quale il farmaco risulta costo efficace per i sistemi sanitari. Il costo previsto potrebbe quindi essere giustificabile solo in un sottogruppo di pazienti in cui l’efficacia sembra maggiore. Il rischio è che, oltre al problema di identificare le persone nella fase di malattia non ancora conclamata attraverso lo studio degli autoanticorpi circolanti e della funzione metabolica, si debbano aggiungere ulteriori caratterizzazioni come ad esempio alcuni assetti genetici. Questo sarà un grosso punto di riflessione più generale per il futuro che dovremo risolvere per evitare il rischio di avere trattamenti potenzialmente efficaci ma non essere in grado di identificare i pazienti che ne possano beneficiare o non poterli sostenere economicamente”.
